About | Downloads | Lists | Workshops | Videos | News | Citations | Icon | Publications
About SLiM
SLiM is a free, open-source evolutionary simulation framework that combines a powerful engine for population genetic simulations with the capability of modeling arbitrarily complex evolutionary scenarios. Simulations are configured via the integrated Eidos scripting language that allows interactive control over practically every aspect of the simulated evolutionary scenarios. The underlying individual-based simulation engine is highly optimized to enable modeling of entire chromosomes in large populations. We also provide a graphical user interface on macOS, Linux, and Windows, for easy simulation set-up, interactive runtime control, and dynamical visualization of simulation output.
A 4–5 day SLiM Workshop is now available online. The SLiM Workshop is also offered in person from time to time; see the SLiM Workshops subsection below for more information.
A podcast called Data Skeptic interviewed Ben Haller about SLiM (23 April 2024); maybe that episode is a good place to start if you’re curious about SLiM? I was also interviewed by the Heredity podcast, associated with the journal Heredity (28 August 2024); you can listen to that interview via Acast or download.
Downloads (version 5.2)
For help with installation on all platforms, see chapter 2 of the SLiM manual. The macOS Installer above will install the slim command-line tool and the SLiMgui graphical development environment. On Linux platforms there may be an installer for your platform, or you can build from sources. On Windows, there is a pacman installer, or you can build from sources; you can also run SLiM on Windows natively or under the WSL.
The SLiM Manual includes a collection of over 200 recipes for common situations. You can download a zip archive of those recipes, if you wish; they are also directly available through SLiMgui’s File menu. The Eidos Manual covers the details of the Eidos language, used to control SLiM via scripting. Reference sheets for both SLiM and Eidos, downloadable above, make them easy to use with minimal use of the full documentation.
Note that the source code archive provided above contains neither macOS specific code, nor the Xcode project for SLiM; it is intended for users on Linux and Windows. The complete sources including macOS files can be found on GitHub; you can get the sources for a tagged release, such as 5.2, or for the current development head.
We also provide a GitHub repository called SLiM-Extras with additional useful tidbits for users of SLiM, such as user-defined Eidos functions for performing some common tasks, and we welcome contributions to that repository from others.
SLiMgui
With the SLiMgui graphical modeling environment (compatible with macOS, Linux, and Windows), you can visualize your simulation as it runs and examine its parameters in real-time, allowing for much easier simulation development.
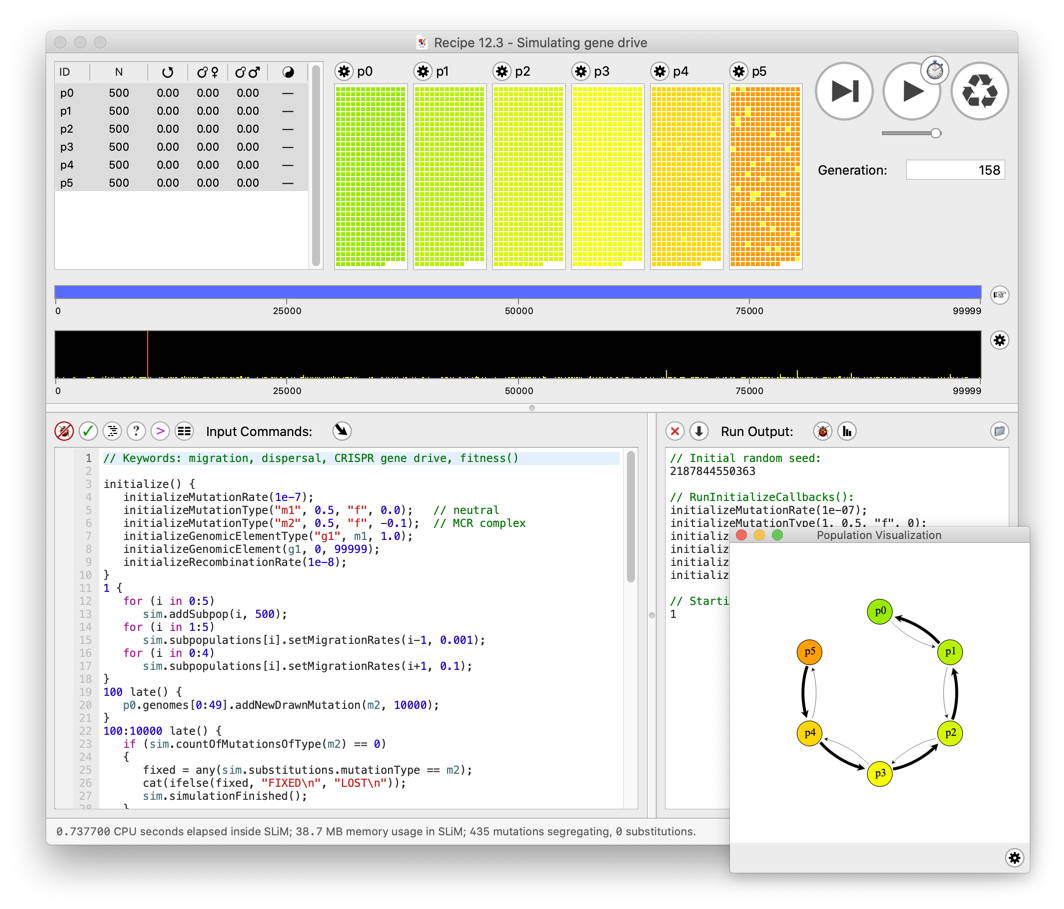
A screenshot of SLiMgui simulating the population dynamics of a CRISPR/Cas9 “gene drive” in a six-subpopulation island model with spatial variation in selection on the driver allele. The Eidos scripting area is on the left, output is on the right. A visual representation of the population structure is shown in the subwindow, and all of the individuals in the six subpopulations can be seen at top (colored according to their relative fitness). The central black bar shows a summary of the genetic variation present in the population; rare neutral mutations are visible as short yellow bars, and the tall red bar represents the driver allele approaching fixation.
Mailing lists and other community resources
There are two mailing lists. Please subscribe to either or both using the links below:
- slim-announce: A low-volume mailing list for announcements only. Users may not post to this list.
- slim-discuss: A higher-volume mailing list for questions from users of SLiM.
A community organization for SLiM users is at https://github.com/slim-community. That is the appropriate place to open new discussions, contribute whole repositories for software related to SLiM, put up job postings related to SLiM, and more.
For guidance on how to file bug reports and feature requests, ask questions, contribute code and example recipes, and otherwise contribute to SLiM, please see the CONTRIBUTING.md file in SLiM’s GitHub repository.
SLiM Workshops
We run 5-day SLiM workshops that are free and open to the public (with registration). The best place to watch for announcements of new workshops is the slim-discuss mailing list. We have one remaining scheduled workshop in Europe for 2026:
- 17 August – 21 August 2026, University of Iceland, Reykjavík, Iceland
And we have one workshop scheduled in North America for 2026:
- 5 October – 9 October 2026, Cornell University, Ithaca, NY, USA
If you would like to host a workshop at your institution, please contact us!
The workshop materials are now online, for people who want to do it themselves; visit the online workshop download page for more information.
An overview of the workshop content
Information on attending SLiM workshops
Downloadable solutions to workshop exercises
Past workshops:
- 15 June – 19 June 2026, University of Tartu, Tartu, Estonia
- 18 May – 22 May 2026, Musée de l’Homme, Paris, France
- 11 August – 15 August 2025, Natural Resources Institute Finland (LUKE), Helsinki, Finland
- 28 July – 1 August 2025, Universität Wien, Vienna, Austria
- 14 July – 18 July 2025, Université de Lausanne (UNIL), Lausanne, Switzerland
- 30 June – 4 July 2025, Université de Rennes, Paimpont, France
- 28 October – 1 November 2024, American Museum of Natural History, NYC, NY, USA
- 10 June – 14 June 2024, University of Edinburgh, Edinburgh, Scotland
- 27 May – 31 May 2024, Marche Polytechnic U. (with U. of Ferrara), Ancona, Italy
- 13 May – 17 May 2024, Globe Institute, Copenhagen, Denmark
- 29 April – 3 May 2024, Centre for Palaeogenetics, Stockholm, Sweden
- 5-9 February 2024, UNAM–Juriquilla, Querétaro, México
- 28 August – 1 September 2023, Harvard University, Cambridge, MA, USA
- 16–20 June 2023, University of New Mexico, Albuquerque, NM, USA
- 6–10 March 2020, University of Iceland, Reykjavík, Iceland
- 13–17 January 2020, Cornell University, Ithaca, NY, USA
- 4–8 November 2019, City University of New York, New York City, USA
- 9–13 September 2019, University of East Anglia, Norwich, UK
- 12–16 August 2019, Umeå University, Umeå, Sweden

The first SLiM workshop: Umeå, Sweden, August 2019.
SLiM videos
We have started making videos related to SLiM, which will be posted here. Note that the SLiM Workshop online materials also include video lectures about SLiM; see the Workshops section for those.
It is strongly recommended that you download these videos rather than streaming them; streaming does not seem to work well for them. If clicking on the link does not start a download (or doesn’t do anything at all), try right-clicking or control-clicking on the link and choosing “Download Linked File” or “Save Link As…” or a similarly-named option from the context menu that perhaps results.
An Introduction to SLiM (Ben Haller): video (1 hour)
13 March 2024: This is one-hour introductory talk about SLiM, first presented during the Modeling & Theory in Population Biology series in 2024. It’s a great place to start if you’re not sure what SLiM is, what it can do, and why you might want to use it.
SLiMgui Power-User Features (Ben Haller):
17 September 2025: This is a three-talk series about using SLiMgui effectively. It focuses mainly on custom plotting in SLiMgui – a really core feature – but introduces lots of useful features along the way.
Part I: video (43 minutes)
This shows how to make a plot of FST over time in a simple neutral two-subpopulation model with vicariance and secondary contact.
Part II: video (21 minutes)
This shows how to make a plot of LD (linkage disequilibrium) along the chromosome, updating each tick as it evolves in a selective sweep model.
Part III: video (31 minutes)
This shows how to make a plot of individuals in a spatial model, showing the relatedness between individuals and grouping them into spatial clusters.
A Practical Introduction to the Tree Sequence (Peter Ralph): video (24 min.)
16 June 2023: This is fairly short talk about the tree sequence data structure, both in the abstract and as used in SLiM simulations. It’ll help you understand what tree sequences are, why you might want to use them, and how to get started with them.
A Walk Through the Gardens of SLiM (Ben Haller): video (1.2 hours)
11 October 2022: This is a look at how SLiM works under the hood, exploring the C++ internals behind SLiM and Eidos. It is not for beginning SLiM users, and it will make more sense if you have a little programming experience.
News
Full release notes for each SLiM release can be found on GitHub at SLiM Releases. Just brief announcement blurbs are now kept here.
2026 April 13: SLiM 5.2 is released! This is a major point release that adds SIMD performance optimizations, new features for SLiMgui custom plotting, and much more. It also has some important bug fixes, and should be used instead of SLiM 5.1.
2025 November 26: Our SLiM 5 paper is published in MBE! B.C. Haller, P.L. Ralph, P.W. Messer. (2026). SLiM 5: Eco-evolutionary simulations across multiple chromosomes and full genomes. Molecular Biology and Evolution 43(1), msaf313. DOI.
2025 October 15: We’ve published a paper on SimHumanity, a model for simulating humanity evolutionary history in SLiM! B.C. Haller, C.W. Nelson, M.F. Rodrigues, P.W. Messer. (2025). SimHumanity: Using SLiM 5.0 to run whole-genome simulations of human evolution. Human Population Genetics and Genomics 5(4), 0006. DOI.
2025 September 13: SLiM 5.1 is released! This is a major point release that adds improved support for matrices/arrays, more pop-gen utility functions, new features for SLiMgui custom plotting, and much more. It also has some important bug fixes, and should be used instead of SLiM 5.0.
2025 April 18: SLiM 5.0 is released! This is a major release that adds support for simulating multiple chromosomes in SLiM. It also has some other nice new features and important bug fixes.
2024 September 17: SLiM 4.3 is released! This is a major point release with some nice new features and important bug fixes. Notably, it shifts SLiMgui from Qt5 to Qt6 for the widget kit that provides its cross-platform functionality.
2024 May 5: SLiM 4.2.2 is released! This is a bug fix release, and should be used instead of SLiM 4.2.
2024 April 12: SLiM 4.2.1 is released! This is a bug fix release, and should be used instead of SLiM 4.2.
2024 March 22: SLiM 4.2 is released! This is a major point release, with a bunch of new stuff, especially including custom plotting in SLiMgui and arbitrary Eidos expressions for event/callback tick ranges.
2023 December 6: SLiM 4.1 is released! This is a major point release, with a bunch of new stuff, especially including new spatial modeling features.
2023 March 21: Our SLiM 4 paper made it out, in the American Naturalist! “SLiM 4: Multispecies Eco-Evolutionary Modeling” describes the new support for multispecies, eco-evolutionary modeling in SLiM 4 (DOI).
2022 September 14: SLiM 4.0.1 is released! This is a minor release, mostly to fix a couple of bugs.
2022 August 12: SLiM 4.0 is released! This is a major release, notably adding support for simulating multiple species including eco-evolutionary dynamics and coevolution.
2022 February 14: SLiM 3.7.1 is released! This is a minor bug-fix release, fixing one crash and several more minor issues.
2021 December 15: SLiM 3.7 is released! This is a major release, with tons of new stuff and several important bug fixes. It adds Windows as a supported platform. This upgrade is recommended for all users, but breaks backward compatibility in a couple of ways for some models.
2021 March 3: SLiM 3.6 is released! This is a major release, with many new features and several important bug fixes. This upgrade is strongly recommended for all users; version 3.5 should not be used due to two bugs that could cause incorrect model results.
2020 December 8: SLiM 3.5 is released! This is a major release, with many new features and several important bug fixes.
2020 May 12: SLiM 3.4 is released! This is a major release, including the first release of QtSLiM, as well as a few other new features and fixes for a couple of bugs.
2020 January 30: SLiM 3.3.2 is released! This is a minor release, with several small improvements and fixes for a couple of bugs (one significant).
2020 January 13–17: We had a SLiM workshop at Cornell, our home turf! We’ve done a couple so far (Sweden, the UK, New York City) but this was our first at Cornell. We had 28 attendees, and it went very well, including a catered lunch sponsored by 3CPG. We’ve got more workshops planned (see the previous subsection); please contact us if you’d like to host a workshop at your institution!
2019 September 28: SLiM 3.3.1 is released! This version is a minor release, mostly bug fixes (including a few rare but nasty bugs!).
2019 May 15: SLiM 3.3 is released! This version is a major release, with big new features (nucleotide-based models, mutation() callbacks, etc.) and some big, important bug fixes.
2019 January 29: SLiM 3.2.1 is released! This version is a minor release, providing some smaller feature additions and a couple of new recipes, as well as bug fixes.
2019 January 18: In recent days we have had three new papers published related to SLiM. (1) “SLiM 3: Forward genetic simulations beyond the Wright–Fisher model” describes the support for non-Wright–Fisher models and continuous space in SLiM 3 (DOI). (2) “Evolutionary modeling in SLiM 3 for beginners” walks new users through making a simple model in SLiM 3, with lots of explanations of basic concepts (DOI). (3) “Tree‐sequence recording in SLiM opens new horizons for forward‐time simulation of whole genomes” discusses the new tree-sequence recording feature in SLiM 3 in detail, with several examples of its practical application to speeding up model execution, burn-in simulation, and data analysis (DOI). We hope these papers are useful for getting up to speed on all the new stuff we’ve been working on!
2018 November 6: SLiM 3.2 is released! This version greatly improves the performance of large nonWF models, and provides numerous improvements and bug fixes.
2018 September 3: SLiM 3.1 is released! This version greatly improves the performance of spatial interactions, and provides improvements to tree-sequence recording, among other improvements and bug fixes.
2018 July 1: SLiM 3.0 is released! This is our first full version upgrade since SLiM 2.0 was released in early 2016. It adds support for non-Wright-Fisher (nonWF) models and tree-sequence recording, two features that greatly increase SLiM’s power and flexibility. Many smaller improvements have been made too.
2017 December 16: SLiM 2.6 is released! This is a major release, with lots of new stuff.
2017 October 27: SLiM 2.5 is released! This is a major release, with lots of new stuff and some important bug fixes.
2017 September 12: SLiM 2.4.2 is released (fix for a bug that could cause incorrect output from many/most models).
2017 July 26: SLiM 2.4.1 is released (fix for a bug that could cause crashes or incorrect results in multi-subpopulation simulations).
2017 July 14: SLiM 2.4 is released! This is a major release, with speed improvements for many types of models, new support for runtime profiling in SLiMgui, and many other features.
2017 April 18: SLiM 2.3 is released! This is a major release, notably adding support for continuous space and spatial interactions.
2017 February 22: SLiM 2.2.1 is released (new recipes, minor features and fixes).
2016 December 8: SLiM 2.2 is released! This is a major release with big performance improvements and several new features. See the new SLiM and Eidos manuals for current documentation. There are also release notes in the announcement on slim-announce.
2016 November 8: SLiM 2.1.1 is released (mostly bug fixes, some serious).
2016 October 4: Our publication on SLiM 2 is out in Molecular Biology and Evolution: DOI.
2016 September 19: SLiM 2.1 is released! This is a major release with lots of features added.
2016 May 26: SLiM 2.0.4 is released (minor bug fixes).
2016 May 12: SLiM 2.0.3 is released (improvements to code completion).
2016 May 6: SLiM 2.0.2 is released (minor feature additions).
2016 April 27: SLiM 2.0.1 is released (minor bug fix and minor feature addition).
2016 April 1: SLiM 2.0 is released! We are excited to announce SLiM 2.0, a new major release of the SLiM package. SLiM 2.0 adds scriptability with Eidos, and interactive simulation development using the SLiMgui application. We have put up a blog post with more details about the SLiM 2.0 release.
License and citation
SLiM is free open-source software, licensed under the GNU General Public License version 3. If you use SLiM in your research, please cite us.
For SLiM 5, cite:
Haller, B.C., Ralph, P.L., & Messer, P.W. (2025). SLiM 5: Eco-evolutionary simulations across multiple chromosomes and full genomes. bioRxiv. DOI
For SLiM 4, cite:
Haller, B.C., & Messer, P.W. (2023). SLiM 4: Multispecies eco-evolutionary modeling. The American Naturalist 201(5), E127–E139. DOI
For SLiM 3, cite:
Haller, B.C., & Messer, P.W. (2019). SLiM 3: Forward genetic simulations beyond the Wright–Fisher model. Molecular Biology and Evolution 36(3), 632–637. DOI
For models using tree-sequence recording, cite:
Haller, B.C., Galloway, J., Kelleher, J., Messer, P.W., & Ralph, P.L. (2019). Tree‐sequence recording in SLiM opens new horizons for forward‐time simulation of whole genomes. Molecular Ecology Resources 19(2), 552–566. DOI
If appropriate, cite our “protocol” paper for beginning SLiM users:
Haller, B.C., & Messer, P.W. (2019). Evolutionary modeling in SLiM 3 for beginners. Molecular Biology and Evolution 36(5), 1101–1109. DOI
For older models using SLiM 2, cite:
Haller, B.C., & Messer, P.W. (2017). SLiM 2: Flexible, interactive forward genetic simulations. Molecular Biology and Evolution 34(1), 230–240. DOI
And for SLiM 1, cite:
Messer, P.W. (2013). SLiM: Simulating evolution with selection and linkage. Genetics 194(4), 1037–1039. DOI
The SLiM icon and QR code
Graphics files for the SLiM icon and QR code are downloadable here. These images are: Copyright (c) 2016–2025 Benjamin C. Haller, All Rights Reserved. Permission is hereby granted for re-use specifically for SLiM-related purposes that are respectful and supportive of the SLiM community. If you have any doubt as to this, please contact us for permission. Various sizes and formats are provided.
Here is the SLiM icon:
![]()
256×256, TIFF format: SLiM_256.tif
256×256, JPEG format: SLiM_256.jpg
1024×1024, TIFF format: SLiM_1024.tif
1024×1024, JPEG format: SLiM_1024.jpg
Vector, PDF format: SLiM_vector.pdf
Here is the SLiM QR code, which redirects to this page:
![]()
656×656, TIFF format: SLiM_Home_QR.tif
656×656, PNG format: SLiM_Home_QR.png
656×656, PNG format with transparency: SLiM_Home_QR_transparent_png
And here is a SLiM icon with its QR code integrated:
![]()
1024×1024, TIFF format: SLiM_with_QR_1024.tif
1024×1024, PNG format: SLiM_with_QR_1024.png
This integrated QR code seems to work for Apple’s QR code recognition system, at least; but other readers might not be as robust, so you ought to test it if you are concerned about possible problems with particular readers.
Finally, here is the SLiM name in our preferred typeface, Hoefler Text:
![]()
8253×2672, PNG format: SLiM_Hoefler.png
This image is rather high-resolution so that it displays smoothly, with minimal aliasing artifacts even at fairly large sizes. If you have Hoefler Text installed on your system, you should simply use that typeface instead.
Older versions
For new projects, using the current version is strongly recommended. Old versions from SLiM 2.0 onward can be found on GitHub using the release tags. These old versions are no longer supported.
SLiM 1.8: documentation source
Publications
It can be useful to see how others are using SLiM; sometimes you can even download the actual SLiM model used in a paper. However, updating a list of all the publications that cite SLiM became too time-consuming, so now we provide links for Google Scholar searches, for papers that cite our SLiM papers.
Citations for SLiM 5 (> 10 cites – brand new!)
Citations for SLiM 4 (> 350 cites)
Citations for SLiM 3 (> 920 cites)
Citations for Tree-sequence Recording in SLiM (> 230 cites)
Citations for SLiM 2 (> 250 cites)
Citations for SLiM 1 (> 230 cites)
