Asymptotic McDonald-Kreitman test
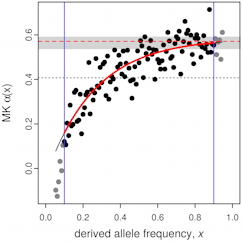 The McDonald–Kreitman test is widely used to estimate the fraction of substitutions in a genomic test region that were driven to fixation by positive selection. However, the original test can be strongly biased by the presence of slightly deleterious mutations. Messer and Petrov (2013) introduced a modified test, the asymptotic McDonald–Kreitman test, which corrects for such biases. Our asymptoticMK web service provides a quick and easy way to run this test in any web browser. It provides both quantitative results from the test and plots such as the one shown here. It is implemented in R using FastRWeb, is open source (link below), and is free to use. It can also be run at the command line as part of an automated workflow; see the asymptoticMK homepage for details.
The McDonald–Kreitman test is widely used to estimate the fraction of substitutions in a genomic test region that were driven to fixation by positive selection. However, the original test can be strongly biased by the presence of slightly deleterious mutations. Messer and Petrov (2013) introduced a modified test, the asymptotic McDonald–Kreitman test, which corrects for such biases. Our asymptoticMK web service provides a quick and easy way to run this test in any web browser. It provides both quantitative results from the test and plots such as the one shown here. It is implemented in R using FastRWeb, is open source (link below), and is free to use. It can also be run at the command line as part of an automated workflow; see the asymptoticMK homepage for details.
Reference:
BC Haller, PW Messer (2017)
asymptoticMK: A web-based tool for the asymptotic McDonald–Kreitman test. G3: Genes, Genomes, Genetics. 7:1569
Gene drive resistance
 This C++ program calculates the frequency trajectory xd(t) of the driver allele under the deterministic model specified by Equations 1-4 in our paper, when assuming absence of resistance alleles. It also calculates the rate uμ(t) at which resistance alleles are expected to arise by de novo mutation, the rate uδ(t) at which they are expected to arise by NHEJ, the effective selection coefficient se(t) of resistance alleles, and the establishment probability π(t) of a resistance allele arising in a single copy in generation t. Results are provided for each generation 0 ≤ t ≤ tfix. Lastly, the individual resistance probabilities Ptot, PSGV, Pμ, and Pδ are reported.
This C++ program calculates the frequency trajectory xd(t) of the driver allele under the deterministic model specified by Equations 1-4 in our paper, when assuming absence of resistance alleles. It also calculates the rate uμ(t) at which resistance alleles are expected to arise by de novo mutation, the rate uδ(t) at which they are expected to arise by NHEJ, the effective selection coefficient se(t) of resistance alleles, and the establishment probability π(t) of a resistance allele arising in a single copy in generation t. Results are provided for each generation 0 ≤ t ≤ tfix. Lastly, the individual resistance probabilities Ptot, PSGV, Pμ, and Pδ are reported.
Reference:
RL Unckless, AG Clark, PW Messer (2017)
Evolution of resistance against CRISPR/Cas9 gene drive. Genetics. 205:827
H-scan
Selective  sweeps elevate levels of linkage disequilibrium (LD) over those observed in neutrally evolving regions and generate unusually long tracts of homozygosity around the adaptive site. This is expected for hard as well as soft selective sweeps, unless they are so soft that the number of distinct adaptive haplotypes in a sample is similar to the expectation under neutrality. The H statistic aims to detect this signature in population genomic data by measuring the average length of pairwise homozygosity tracts along the genome in a population sample. H-scan is an easy-to-use C++ command line program.
sweeps elevate levels of linkage disequilibrium (LD) over those observed in neutrally evolving regions and generate unusually long tracts of homozygosity around the adaptive site. This is expected for hard as well as soft selective sweeps, unless they are so soft that the number of distinct adaptive haplotypes in a sample is similar to the expectation under neutrality. The H statistic aims to detect this signature in population genomic data by measuring the average length of pairwise homozygosity tracts along the genome in a population sample. H-scan is an easy-to-use C++ command line program.
H-scan is distributed under the GNU General Public License (GPL)
Indel trace extension
The indel trace extension method evaluates whether an insertion of a sequence segment is a duplication of an adjacent sequence segment, rather than just a random piece of DNA. Furthermore, it allows to analyze whether duplicates were already present at the insertion site before the duplication event occurred. Likewise, the method can detect whether deletion events removed one copy of a preexisting duplicate.

Fig: Illustration of the trace extension method. The top part of the figure shows the dot-matrix of an indel. Different relations between the indel length l and its trace extension d distinguish four different classes of events shown in the bottom part of the plot.
Reference:
PW Messer, PF Arndt (2007)
The majority of recent short DNA insertions in the human genome are tandem duplications. Mol Biol Evol. 24:1190
CorGen
Long-range correlations in DNA are characterized by a power-law decaying autocorrelation function of the sequence composition. Given a DNA sequence as input, CorGen can measure its composition correlation function and determine amplitude and decay exponent of present long-range correlations. The obtained parameters can then be used to generate random sequences with the same correlation parameters and average sequence composition as the query sequence. CorGen can also generate sequences with user specified long-range correlations and GC-content.
Reference:
PW Messer, PF Arndt (2006)
CorGen — measuring and generating long-range correlations for DNA sequence analysis. Nuc Acid Res. 34:W692
